

Getting mission critical medical products launched to market means getting FDA approval. And these days that process often starts in software development. The Food and Drug Administration regulates the medical device market, with many constraints on developing products.
First, it may be helpful to talk about the history of the FDA and its importance in today’s medical-device market.
The History of the FDA
When the FDA was born, it bore no similarity to the version you know today. The administration only chased after companies that already had devices or medications in the market that were proven to harm patients. Its mandate wasn’t preventing harmful material from being launched into the market. For example, in the 1930s, you could buy a radium belt that consisted of radioactive materials.
1970s
As products proliferated and technology grew, the FDA finally realized it needed to regulate the industry due to two prominent device failures:
- The pacemaker: 10% of pacemakers failed in the first month after implantation; 50% in first three months
- The IUD: Intrauterine devices were harming women to the point where they were becoming infertile. IUDs also caused infections to hundreds of thousands of women who were harmed by unsafe and ineffective products.
So, you can see why the FDA started monitoring the quality of devices before they launched to market. In 1976, the FDA’s Medical Device Regulation Act addressed the safety of devices with two rules:
- All medical-device manufacturers must get approval before going to market with a medical device.
- If the manufacturer could prove that a product was substantially equivalent to a product already on the market before 1976, it could get faster approval from the FDA.
1990s
In the 1990s, the FDA realized companies were using software to manage manufacturing, release products, create electronic signatures, and maintain databases. The FDA was used to getting this information on paper; the electronic equivalents were fallible and no one at the FDA trusted them.
This led to the FDA passing two regulations that required companies, as part of proving a product was safe and releasing it to the market
What the FDA Does Today
At the beginning of the twenty-first century, developers started developing software-only medical products. These flew under the radar of the FDA because no device or hardware was being manufactured and these software products didn’t physically have any contact with patients. They became known as either software as medical devices (SaMDs) or mobile medical applications (MMAs).
Eventually, the FDA started taking notice and drafted guidelines to help manage this software. Several regulations were drafted for different categories
- Medical device software: A medical device that interacts with a patient.
- Production software: Any software used to manufacture a device.
- Quality system software: Software used to release a device.
- Mobile medical apps: Apps installed on a phone; not dedicated hardware.
- Software medical device: Apps installed elsewhere; for example, desktop or the web.
- Medical device data system: Used to transfer data from one device or app to another.
Each category can be broken down further. For example, software medical devices fall into three classes:
- Class 1: This class includes general controls; for example, tongue depressors. You don’t need to wait for approval, you just have to prove that they aren’t harmful—the tongue depressors aren’t moldy or full of lead.
- Classes 2 & 3: These classes require special controls and pre-market approval by the FDA. The devices in these classes are those in which a medical professional makes decisions based on the device or those that can physically cause harm. An example of a class 2 device is a blood-glucose monitor. Class 3 is any device transplanted in the body that could cause harm; for example, an infusion pump or an artificial heart.
CFR 21 Part 820 Design Controls
CFR 21 Part 820 Design Controls are the guidelines for how to design and develop Class 2 and Class 3 medical devices for use in the US market.
Notice I didn’t say “rules.” The FDA doesn’t issue absolute rules—only guidelines to show that you did your due diligence that your device won’t cause harm once it’s on the market. However, you do need to show that you followed a quality policy, including management reviews and audits.
Developing Devices
Everyone’s goal is to build a high-quality product that won’t hurt people. But no matter how careful or thoughtful developers are, bugs always exist in products. No devices would ever enter the market if they needed to be bug free—they would be in an endless development cycle. Fortunately, the FDA understands that.
The FDA asks that you take a risk-based approach when developing medical devices. This approach allows you to focus your effort on areas of the product that are more critical and where the potential to cause harm is higher. You still design and test every area of your product or device, but you spend the majority of your time on those critical areas.
This approach entails showing the FDA that you thought through what you’re building and that you’re testing to ensure quality. How are you going to test it? Have you independently tested it (the person building it wasn’t the person testing it)? Have you documented every step? Documentation is very important to the FDA to show evidence of following the process.
There are several testing models you can use.
Testing to Ensure Quality
The classic testing model is the V-Model (or Verification and Validation model). You create user needs, design input, test that input, verify that the output meets your design, and at the end you have a product that you can then validate.
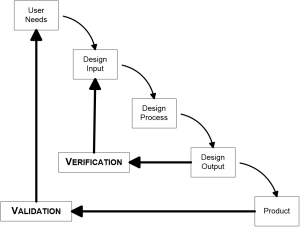
The drawback to the V-Model is its waterfall effect. You design everything up front, and then test it. If one thing goes wrong, you must go back to redo the entire process. Documentation for the V-Model is complicated because you have to show the FDA why you need to go back and redo. And you will get questioned why you needed to make changes in an audit of your product.
Even though developers use V-Model, the FDA actually doesn’t require a waterfall type of model. Its usefulness in practice is limited, and for more complicated devices or products a concurrent model of testing is more useful.
Which is where the agile model steps in. A lot of the aspects of the agile method ensure quality and are actually well-aligned with the FDA guidelines. You still go through the design/build/verify loop; you can just get through those loops quicker. The idea is that you split your projects into chunks and go through the design/build/verify loop for each chunk. You can then be confident of each chuck before you move on to the next.
The agile testing quadrants support verification and validation.
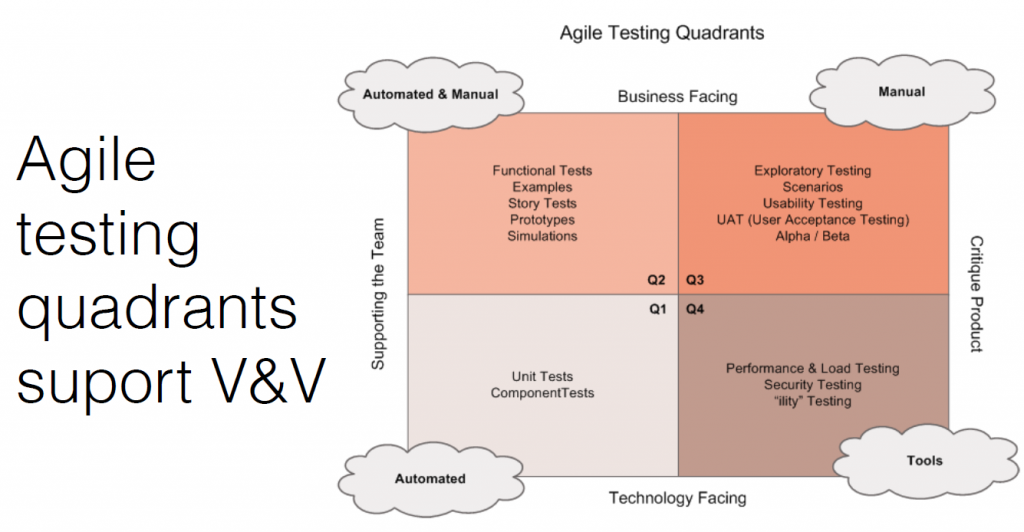
- Internal verification testing: This includes stories, manuals, functional tests, and prototypes. How you show work is done and meets the pre-defined objectives.
- External manual testing: This is testing in front of users, and seeing how they use and misuse the product. Testing with lots of people in different types of situations is important.
- Internal automated testing: Achieved through unit and integration tests with a target code coverage metric (typically > 90%)
- Tools: This includes performance, security, load testing, and penetration testing. The FDA likes to see a broad coverage of testing.
If you’re not sold on agile development yet, it has other useful aspects for software development:
- It allows you to focus on flow. You don’t have everyone working on everything at once and hoping everything comes together.
- Your focus can be limited to a specific chunk in progress.
- You can incorporate test-driven development. You know what the acceptance criteria is before you start development which helps developers know when they’re done.
- You can perform more valuable code reviews. Someone can look at each release or task worth of code in manageable chunks making code reviews more valuable and targeted.
- You can take advantage of pair programming. You can have someone double check work, which is also built in security. Multiple people understand how the code works (shared understanding). If someone leaves the project or the company, you’re not left with code no one understands.
- It allows for continuous integration. You can make sure everything is working together continuously to catch bugs early and often, rather than at the end.
- Building automated tests as you go is a valuable tool for evaluating the quality of your code iteratively over time that results in a very robust regression testing tool that can live with the software.
Technology
As long as you use technology the FDA understands, you can use whatever technology you want. If you’re using new and experimental technology the FDA doesn’t understand (such as artificial intelligence), expect to spend some time educating the FDA and for the process to take longer before you get approval.
Security
Security is important for every device, but equally—and maybe even more important—for medical devices. Security holes leave patients vulnerable to outside influences or influencers.
One well-known case is from Johnson & Johnson’s insulin pump with a controller. There was absolutely no encryption built in between the pump and controller, which allowed the key to be available to anyone. There was no protection from relay attacks. A hacker could insert bad code and strings and replay it back to the pump.
The advice that you get from the consumer market is applicable to the medical-device field. For example, account setup, password control, and authenticating. Don’t overlook the security aspect of your software.
Conclusion
Other things you’re exposed to in the medical device development field:
- Document all the details of the design and development. You must document every step you’ve taken and maintain that documentation through 10 years after the lifetime of your device or product. Use technology to your advantage here. If an automated documentation tool can output an HTML report for a task in a human-readable format, use it! Automatically generating a report or using self-documenting APIs can save a lot of time.
- Validate your tools and software. You need to prove that the tools you’re using can be trusted to verify your software. Lock in versions and dependencies of your tools and document them. If you upgrade, you’ll need to retest, and re-document.
- Be prepared to be patient. FDA approvals can take years. And you may see your product or device never getting approved.
- Be innovative, but not too innovative. You can experiment in controlled, approved environments—not in the market with live people.